

Introduction
Gene therapies have transformed treatment for devastating diseases by offering one-time, potentially curative therapies where few or no effective alternatives exist. While acknowledging this transformation in medicine, the healthcare industry has also had to grapple with how to balance the meaningful benefits of gene therapies with their substantial cost. With treatment costs often reaching into the millions of dollars, payers must carefully consider utilization management strategies that limit financial exposure while preserving access for the patients most likely to meaningfully benefit from gene therapy. How has this played out in real-world coverage policies, and are there learnings from the first gene therapy approvals that can be applied to pipeline programs to streamline downstream market access considerations once approved?
To answer these questions, Triangle Insights analyzed payer coverage across several recently-approved gene therapies using Policy Reporter’s payer data and policy platform. Key coverage criteria was compared to the pivotal clinical trial design of these gene therapies in order to better understand how clinical development can shape downstream commercial access. Overall, coverage policies tend to reflect the eligible patient populations that are characterized by the inclusion and exclusion criteria within the pivotal trial design. Coverage then may either be expanded beyond that criteria, or additional criteria may be added, based on available treatment options and the relative unmet need within the condition. Based on this evaluation, we identified three “archetypes” of gene therapies with different access characteristics: 1) gene therapies in indications with the highest degree of unmet need and no alternative treatment options 2) gene therapies in indications with high unmet need but with existing, though inadequate, non-gene therapy treatment options and 3) gene therapies in indications with relatively adequate non-gene therapy treatments (Figure 1).
For gene therapies in indications with the highest unmet need and no meaningfully effective alternative treatments (Archetype 1), clinical trial eligibility criteria and payer access restrictions are both typically limited, with requirements primarily focused on confirming diagnosis and disease stage. In contrast, in indications where adequate non-gene therapy treatment options are available, payers may impose additional restrictions to focus access on patients who remain underserved by the standard of care (Archetype 3). Taken together, these archetypes highlight that payer access for gene therapies is not uniform but instead varies meaningfully based on the level of unmet need and the availability of adequate treatment alternatives.
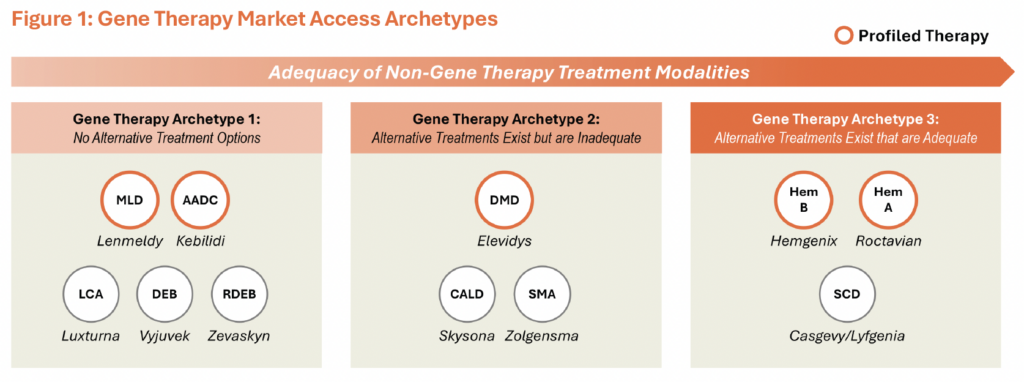
MLD: Metachromatic Leukodystrophy; AADC: Aromatic L-Amino Acid Decarboxylase (AADC) Deficiency; DEB: Dystrophic Epidermolysis Bullosa; RDEB: Recessive Dystrophic Epidermolysis Bullosa; LCA: Leber Congenital Amaurosis; CALD: Cerebral Adrenoleukodystrophy; SMA: Spinal Muscular Atrophy; DMD: Duchenne Muscular Dystrophy; SCD: Sickle Cell Disease; Hem B: Hemophilia B; Hem A: Hemophilia A
Archetype 1: Gene Therapies in Indications with the Highest Unmet Need and No Effective Non-Gene Therapy Options
In conditions with the highest level of unmet need, trial eligibility criteria and associated coverage policies may primarily serve to confirm diagnosis and disease stage, as there is no effective non-gene therapy through which to further refine access. Kebilidi for aromatic L-amino acid decarboxylase (AADC) deficiency and Lenmeldy for metachromatic leukodystrophy (MLD) are examples of this group of gene therapies.
Kebilidi
AADC deficiency is a devastating condition that, untreated, causes severe neurological impairment, motor dysfunction, and, in many cases, early death. Before the launch of Kebilidi in 2024, treatment was limited to symptom management (e.g., dopamine agonists and MAO inhibitors) that produce moderate and inconsistent benefit, leaving patients will little meaningful disease control.
In the absence of effective non-gene therapy treatment options, Kebilidi’s trial participation criteria remained broad, with the primary eligibility criteria including genetically confirmed AADC deficiency with typical clinical characteristics and reduced plasma AADC enzyme activity. Additional requirements such as “persistent neurological symptoms… despite standard therapy” or inability to ambulate independently1 are characteristic of most patients with AADC deficiency and therefore did not materially limit eligibility. As such, the pivotal trial criteria for Kebilidi both reflected the disease’s poorly controlled nature and allowed inclusion of the majority of individuals with genetically confirmed AADC deficiency.
An analysis of major payer policies representing ~50 million covered lives demonstrated that payers closely aligned coverage policies to the participation criteria of Kebilidi’s pivotal trial. With no effective alternative treatments available, payers have not imposed additional access restrictions beyond trial-based eligibility criteria, and as such, these coverage policies effectively allow most diagnosed patients to qualify for treatment. In some instances, payer policies reflect even broader eligibility for Kebilidi than the participation criteria for the trial itself (e.g., no requirement that patients are unable to ambulate independently), further underscoring the role of high unmet need in driving coverage simply tied to confirmed diagnosis.
Lenmeldy
Similar to AADC, MLD is a rapidly progressive, neurodegenerative disorder that results in severe disability (e.g., motor dysfunction, cognitive decline) and, often, early death. Prior to Lenmeldy’s 2024 launch, patients had few, if any, effective therapeutic options, and management was largely limited to supportive and palliative care.
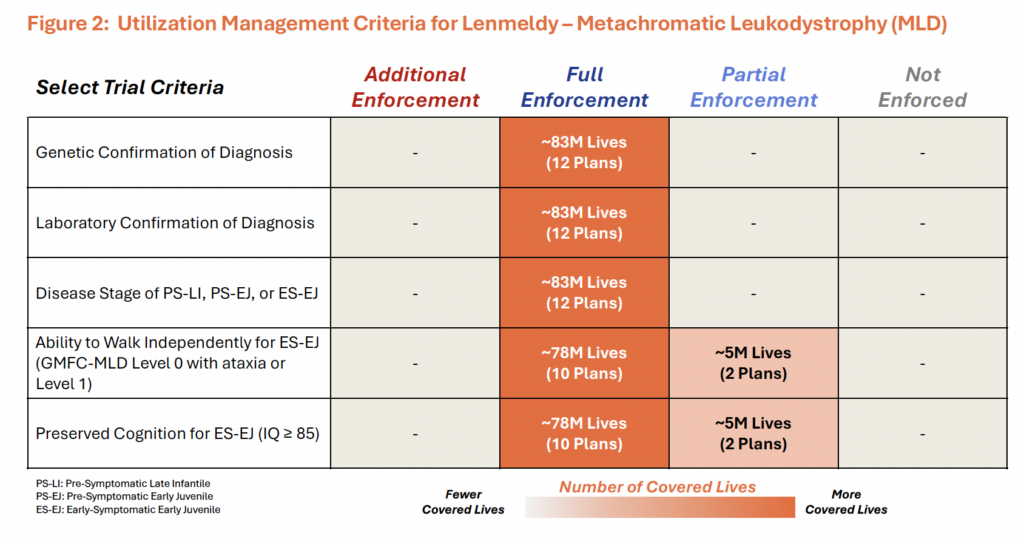
Lenmeldy’s pivotal trial reflected the severity of unmet need by broadly including patients who could still benefit from treatment. Given that Lenmeldy halts but does not reverse disease progression, eligibility was focused on patients treated early in the disease course. Patients were grouped into three broad categories: 1) pre-symptomatic late-infantile (PS-LI MLD) with expected onset ≤30 months, 2) pre-symptomatic early juvenile (PS-EJ MLD) with expected onset between 30 months – 7 years and 3) early-symptomatic early juvenile (ES-EJ MLD). Among these groups, only the early-symptomatic early juvenile patients faced additional eligibility requirements beyond qualifying ARSA genotype, including preserved cognitive function and the ability to walk independently. These criteria were likely intended to ensure treatment was administered early enough in the disease course for patients to derive meaningful clinical benefit.
Payer policies for Lenmeldy have closely mirrored the trial criteria, including the broadly defined eligible patient groups (figure 2). In the absence of effective non-gene therapy treatment options through which to refine access, the diagnostic criteria (e.g., confirmed ARSA mutation and corresponding reduced ARSA enzymatic activity) allow most patients to qualify for coverage of Lenmeldy. Moreover, a few policies (~5M covered lives) did not fully enforce the ambulation and cognitive criteria applied in Lenmeldy’s clinical trial, suggesting that in this archetype, coverage can at times be even broader than the pivotal study population. Together, as with Kebilidi, Lenmeldy’s broad trial criteria and aligned coverage policies underscore how high unmet need can support broad payer access.
Archetype 2: Gene Therapies in Indications with Existing but Inadequate Non-Gene Therapy Treatment Options
For gene therapies in Archetype 2, which target indications with existing but inadequate non-gene therapy options, payers may recognize meaningful remaining unmet need and support use of both gene therapy and non-gene therapy treatment modalities, provided the pivotal trial demonstrates benefit in patients already on conventional therapy. Elevidys in Duchenne muscular dystrophy (DMD) provides a real-world example of this dynamic.
Elevidys
Unlike AADC and MLD, DMD reflects a more nuanced unmet need profile, where exon skipping therapy represented the first major wave of therapeutic innovation and delivered meaningful clinical benefit, yet significant residual unmet need remained. Not all DMD patients are eligible for treatment with exon skipping therapy, and even among eligible patients, these therapies enable production of functional but truncated dystrophin at levels well below typical levels of dystrophin in people without DMD2,3. Further, uptake of exon skipping therapy in cardiac muscles is limited, leaving an important gap in addressing cardiomyopathy, the leading cause of death in DMD4.
Elevidys’ pivotal trial reflects this therapeutic landscape by including both treatment-naive patients and those receiving exon skipping therapy. This trial design allowed the manufacturer to demonstrate broad clinical value, including benefit among patients already treated with exon skipping therapy. As a mutation-agnostic gene therapy, Elevidys expanded the addressable DMD population and showed strong efficacy through higher dystrophin expression and improved penetration into cardiac tissue5. Moreover, the one-time administration of Elevidys could reduce the treatment burden relative to exon skipping therapy, which require lifelong weekly IV infusions. Together, this clinical trial design positioned Elevidys not as an alternative to exon skipping therapy, but as therapy with additive benefit.
Payer coverage of Elevidys has largely aligned with this trial design, with policies typically allowing patients receiving exon skipping therapy to be treated with Elevidys. Given the design of Elevidys’ clinical trial, which demonstrated additive benefit to exon skipping therapy, payers have not included requirements of poor disease management on exon skipping therapy prior to treatment with Elevidys. Elevidys therefore illustrates how thoughtful clinical trial design can support broad access enabling commercial success even in indications with alternative disease-modifying therapies.
Archetype 3: Gene Therapies in Indications with Adequate Non-Gene Therapy Treatment Options
For therapies in the third archetype, the existence of effective non-gene therapy treatment options provides another lever for payers to tailor reimbursement criteria to patients who are not adequately controlled on existing therapies, often extending utilization management criteria beyond clinical trial eligibility criteria. Hemgenix and Roctavian illustrate this dynamic, with payer coverage for both therapies commonly tied to inadequate control on existing therapies. Although Hemgenix demonstrates that therapies in this archetype may still be commercially viable, Roctavian illustrates how a lower degree of remaining unmet need can contribute to more limited uptake and commercial sustainability.
Roctavian and Hemgenix
Roctavian and Hemgenix are gene therapies approved for the treatment of hemophilia A and hemophilia B, respectively, which are inherited bleeding disorders caused by deficiencies in key clotting factors. For both hemophilia A and hemophilia B, treatment traditionally included prophylactic factor replacement, though additional non-factor prophylactic treatments are now available for hemophilia A (e.g., Hemlibra). Compared with prophylactic factor replacement, which generally requires intravenous infusion, Hemlibra offers a simpler subcutaneous route of administration and efficacy comparable to or greater than Factor VIII prophylaxis6, further reducing the remaining unmet need in hemophilia A relative to hemophilia B.
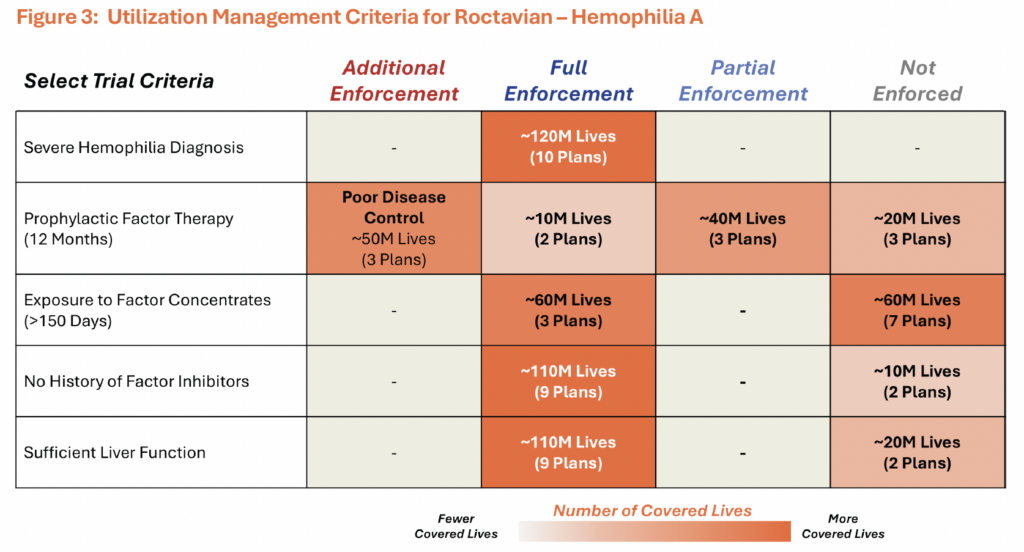
In their pivotal trials, both Hemgenix and Roctavian had relatively broad eligibility criteria centered on adult age, confirmed diagnosis, absence of inhibitors, and prior prophylactic factor replacement therapy, though there was no explicit requirement that patients experience inadequate disease control on factor replacement therapy.
Although pivotal trial eligibility criteria for Hemgenix and Roctavian were relatively broad, analysis of plans covering ~125 million U.S. lives shows that, unlike in higher-unmet-need indications, payers often apply utilization management criteria that extend beyond those trial requirements. For Roctavian, prior to discontinuation, payer coverage criteria generally aligned with core trial-based requirements, however, across the assessed policies, plans representing ~40% of the covered lives evaluated also required inadequate control despite prophylactic or on-demand therapy (Figure 3). This may have represented a meaningful access hurdle, as extended half-life factor VII prophylaxis, such as Altuviio, is associated with zero bleeds in more than half of treated patients7. Similar patterns were observed for Hemgenix across assessed plans.
Despite broadly similar utilization management, Hemgenix and Roctavian have had very different commercial outcomes. Hemgenix has remained on the market, with CSL anticipating growing uptake8, while BioMarin voluntarily withdrew Roctavian from the market in February 2026 after failing to find a buyer. This divergence likely reflects more than payer coverage alone and points to the importance of remaining unmet need. While prophylactic clotting factor replacement is available for both disorders, the availability and efficacy of Hemlibra significantly reduces the remaining unmet need in hemophilia A relative to hemophilia B. A similar dynamic is evident in hemophilia B, where Beqvez entered as a second-to-market gene therapy after Hemgenix had already reduced the remaining unmet need, substantially narrowing the commercial opportunity.
Conclusions and Future Considerations
This evaluation demonstrates that payer access to gene therapy is shaped not only by the clinical profile of the therapy, but by the degree of remaining unmet need in the target indication. Across the therapies assessed, pivotal trial eligibility criteria commonly serve as the foundation for payer coverage policies; however, the extent to which payers broaden or further restrict access varies meaningfully by archetype. In indications with the highest unmet need and no effective alternatives, such as AADC deficiency and MLD, payers largely mirror broad trial criteria and focus coverage on confirming diagnosis and treating patients early enough to benefit. In contrast, where effective non-gene therapy options exist, payers more frequently impose additional utilization management requirements, such as inadequate disease control on standard therapy, to ensure gene therapy is reserved for patients with clear residual need.
Looking ahead, the pricing and access landscape for gene therapies is likely to evolve as new therapies are developed and marketed for the treatment of non-rare diseases (e.g., REGENXBIO’s RGX-314 for the treatment of wet AMD9 and diabetic retinopathy, MeriaGTX’s AAV-GAD for the treatment of Parkinson’s disease10, and Kriya’s KRIY-869 for the treatment of Type 1 diabetes11). As these higher-cost therapies are approved for broader populations, payers are likely to impose more strict utilization management criteria to focus access on patients with the highest anticipated clinical benefit. Strategic clinical trial design may therefore become increasingly important to identify and demonstrate benefit in subpopulations with meaningful remaining unmet need, where access criteria may be less stringent compared to the broader indication.
Beyond indication-specific access criteria, gene therapies targeting broader populations may also face growing pressure from policy-driven pricing reforms, including Inflation Reduction Act (IRA)-related Medicare pricing negotiations and Most Favored Nations (MFN)-related international pricing dynamics. For gene therapies targeting Medicare-heavy populations (e.g., wet AMD, diabetic retinopathy), IRA-related pricing negotiations may increase pressure to demonstrate durable and differentiated clinical value, as negotiated pricing is influenced in part by the therapy’s benefit relative to available alternatives. MFN-related international pricing dynamics may compound this pressure by narrowing pricing flexibility, particularly in non-rare indications, where lower prices in ex-U.S. markets with greater sensitivity to budget impact may place downward pressure on U.S. pricing. Together, these policies may increase the burden on manufacturers to demonstrate a differentiated value proposition.
Having a clear understanding of likely access dynamics can also help to inform priorities within a broader launch strategy. To position gene therapies competitively across indications with varying levels of unmet need, launch investments can be tailored to address the barriers most likely to limit uptake. For gene therapies launching into “Archetype 1” markets, investment may be more heavily prioritized towards patient identification and diagnosis, as access may be less of a hurdle relative to other considerations. For gene therapies entering “Archetype 3” markets, manufacturers may need to place greater priority on access and access support services. For programs earlier in development, understanding these dynamics early can also help to inform clinical development that ultimately, if approved, enables access for patients who can benefit the most.
References
1. Pearson, T., et al., (2020). AADC deficiency from infancy to adulthood: Symptoms and developmental outcome in an international cohort of 63 patients. Journal of Inherited Metabolic Disease, 43(5), 1121–1130.
2. Leckie J., et al., An Updated Analysis of Exon-Skipping Applicability for Duchenne Muscular Dystrophy Using the UMD-DMD Database. Genes. 2024;15(11):1489.
3. Filonova G., et al., Next steps for the optimization of exon therapy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2023;23(2):133–143.
4. Etxaniz U., et al., AOC 1044 induces exon 44 skipping and restores dystrophin protein in preclinical models of Duchenne muscular dystrophy. Nucleic Acids Res. 2025;53(6):gkaf241.
5. Mendell JR., et al., Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-Dystrophin in Children With Duchenne Muscular Dystrophy: A Phase 1/2a Clinical Trial. JAMA Neurol. 2020;77(9):1122–1131.
6. Mahlangu J., et al., Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. 2018;379:811-822.
7. von Drygalski A., et al., Efanesoctocog Alfa Prophylaxis for Patients with Severe Hemophilia A. N Engl J Med. 2023;388:310-318.
8. CSL Company Press Release. CSL announces rise in full year net profit. August 18, 2025.
9. RegenX Bio Company Website. Therapeutic Pipeline. https://www.regenxbio.com/therapeutic-programs. Accessed April 2026.
10. MeiraGTx Company Website. Programs and Pipeline. https://meiragtx.com/programs-pipeline/parkinsons-disease. Accessed April 2026.
11. Kriya Therapeutics Company Website. Pipeline. https://kriyatherapeutics.com/pipeline. Accessed April 2026.